
Case of the Month
October 2017 - Presented by Dr. Nima Amini (Mentored by Dr. John Paul Graff)
Diagnosis
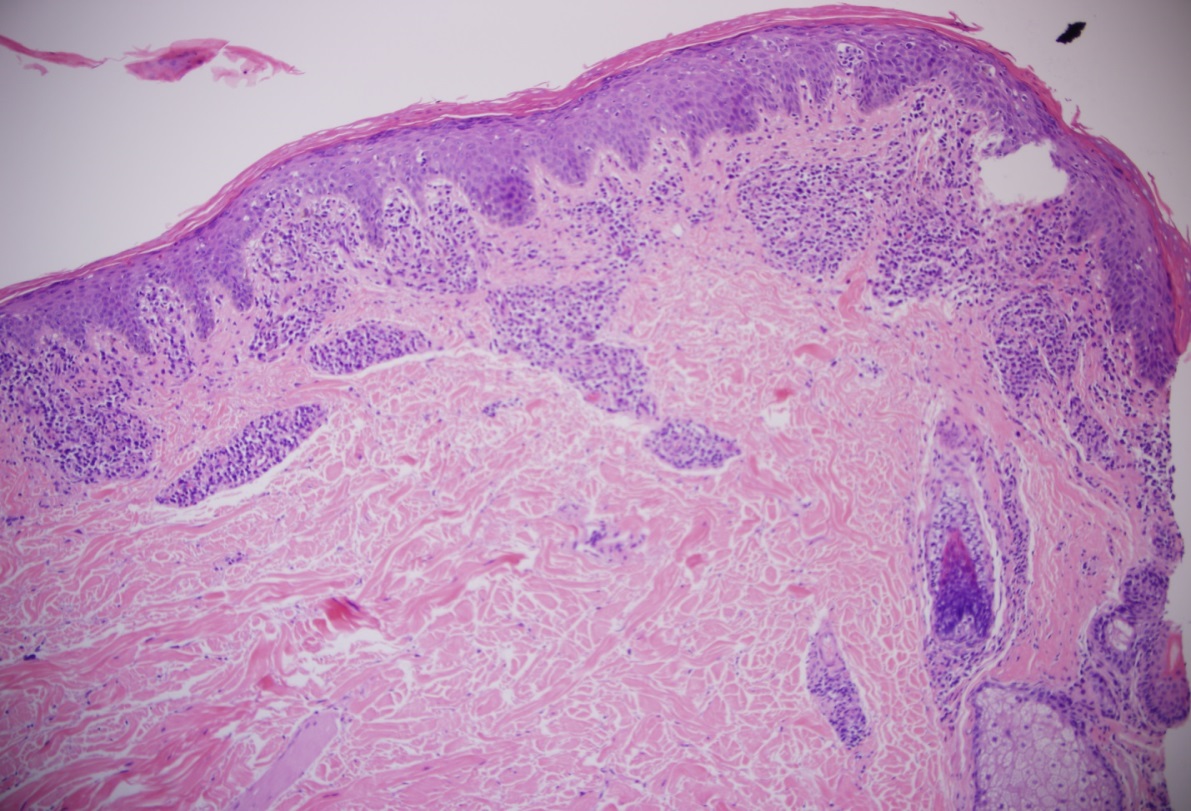
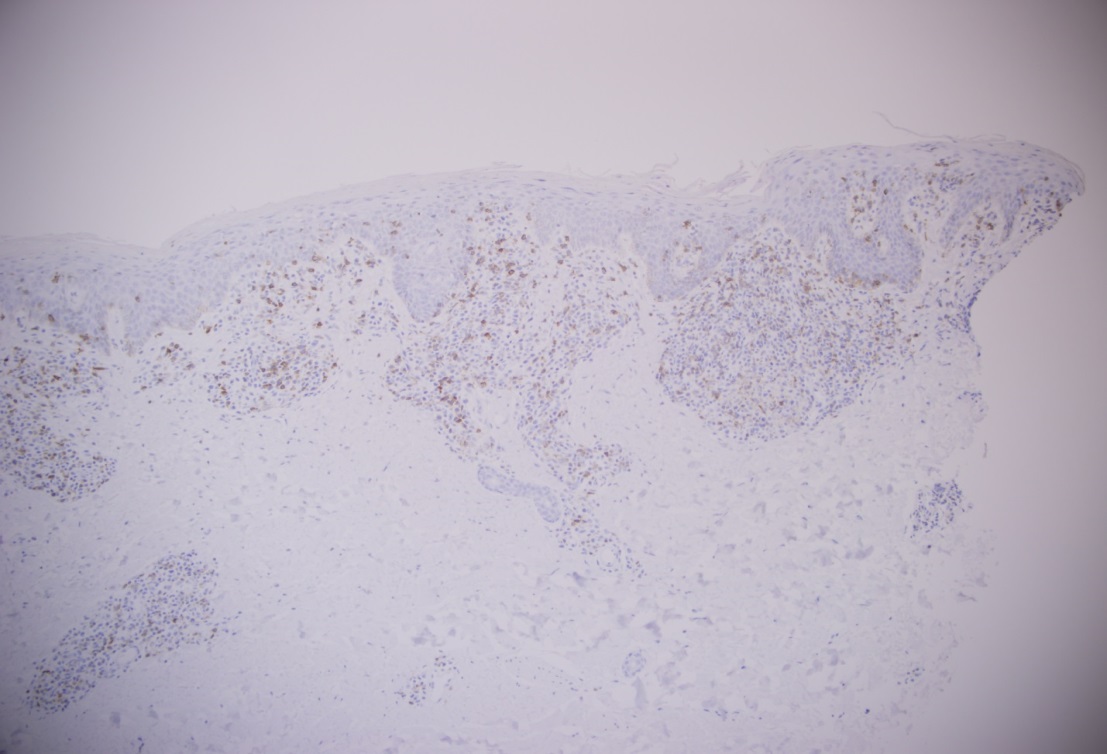
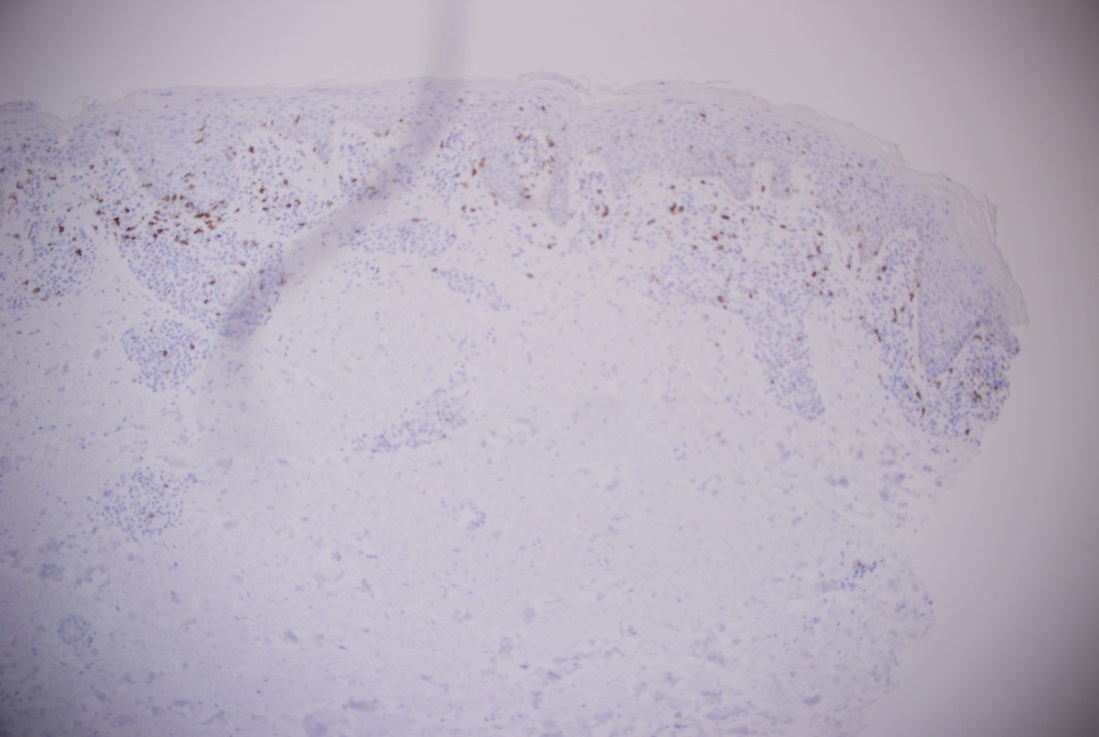
Although the patient has a history of bullous pemphigoid, sections of the specimen from the right palm biopsy showed atypical lymphoid infiltrate with focal epidermotropism. A feature that is not seen in in bullous pemphigoid. A biopsy performed on the right upper arm showed similar findings (figures 4 and 5). Immunohistochemistry (figures 6-9) was performed on the specimen from the right upper arm which showed a superficial perivascular, periadnexal, and focally epidermotropic CD3-positive, CD4-positive, CD8-negative atypical T-cell infiltrate. CD7 expression appeared reduced. Interestingly this immunophenotype matched with the immunophenotype of the T-cell population found in the peripheral blood flow cytometry study. Overall, findings are consistent with a CD4+ T-cell lymphoproliferative process.
The differential diagnoses for our patient includes:
- Cutaneous involvement of T-cell prolymphocytic leukemia
- Adult T-cell leukemia/lymphoma
- CD4-positive cutaneous lymphomas, including mycosis fungoides or Sezary syndrome
T-cell prolymphocytic leukemia (T-PLL) is an aggressive T-cell leukemia. T-PLL is very uncommon and represents approximately 2% of cases of mature lymphocytic leukemias in adults over the age of 30. Skin infiltration is seen in 20% of patients.
Cutaneous involvement consists of perivascular or more diffuse dermal infiltrates without epidermotropism. T-PLL usually presents with WBC count of more than 100K/MM3. Splenomegaly is commonly present and lymphadenopathy is seen in some cases. In the peripheral blood large cells with distinct central nucleoli (prolymphocytes) are seen. Sometimes small cells without nucleoli (often described as indistinct or inconspicuous) are seen. In more than 60% of cases a T-helper phenotype (CD3+, CD4+, CD8-) is seen. Also, in some cases aberrant deletion of one or more T antigens can be seen. In approximately 20% of cases, CD4/CD8 co-expression is seen (common in small-cell variant). A small percentage (10-15%) show cytotoxic phenotype (CD3+, CD4-, CD8+). TCL-1 expression is seen in 70% of the cases. It is often associated with inv(14)(q11;q32) but other cytogenetic deficiencies such as trisomy of the long arm of chromosome 8 can also be seen (per WHO 2008).[1,2]
Adult T-cell leukemia/lymphoma associated with Human T-Lymphotropic Virus 1 is usually divided into four clinical subtypes: acute, chronic, smouldering and lymphomatous. It has some clinical overlap with mycosis fungoides/Sezary syndrome. HTLV-1 studies are needed to confirm the diagnosis. Bizarre flowerlike, (floret) lymphocytes are seen in the peripheral blood. Cells usually show T-helper phenotype, CD3+, CD4+, CD8-. They are generally CD7- and CD25+. Clonal integration of HTLV-1 is seen. Clonal cytogenetic abnormality may also be present.[2]
Sezary syndrome is the leukemic phase of mycosis fungoides (cutaneous T-cell lymphoma) which is defined by the diagnostic triad:
- Diffuse erythroderma
- Lymphadenopathy
- Circulating lymphoma cells (Sezary cells)
In peripheral blood, Sezary cells are distinctly abnormal, convoluted (cerebriform) and usually larger than background normal lymphocytes. Typically, they have T-helper immunophenotype with deletion of expression of CD7 (CD2+, CD3+, CD4+, CD5+, CD7-, CD8-). These cells also show variable or absent expression of CD25 (IL-2 receptor). Clonal cytogenetic deficiencies are common but no single abnormality is characteristic.[2]
Click on figure to enlarge.
|
Figure 4 |
 |
|
Figure 5 |
|
|
|
Figure 6 |
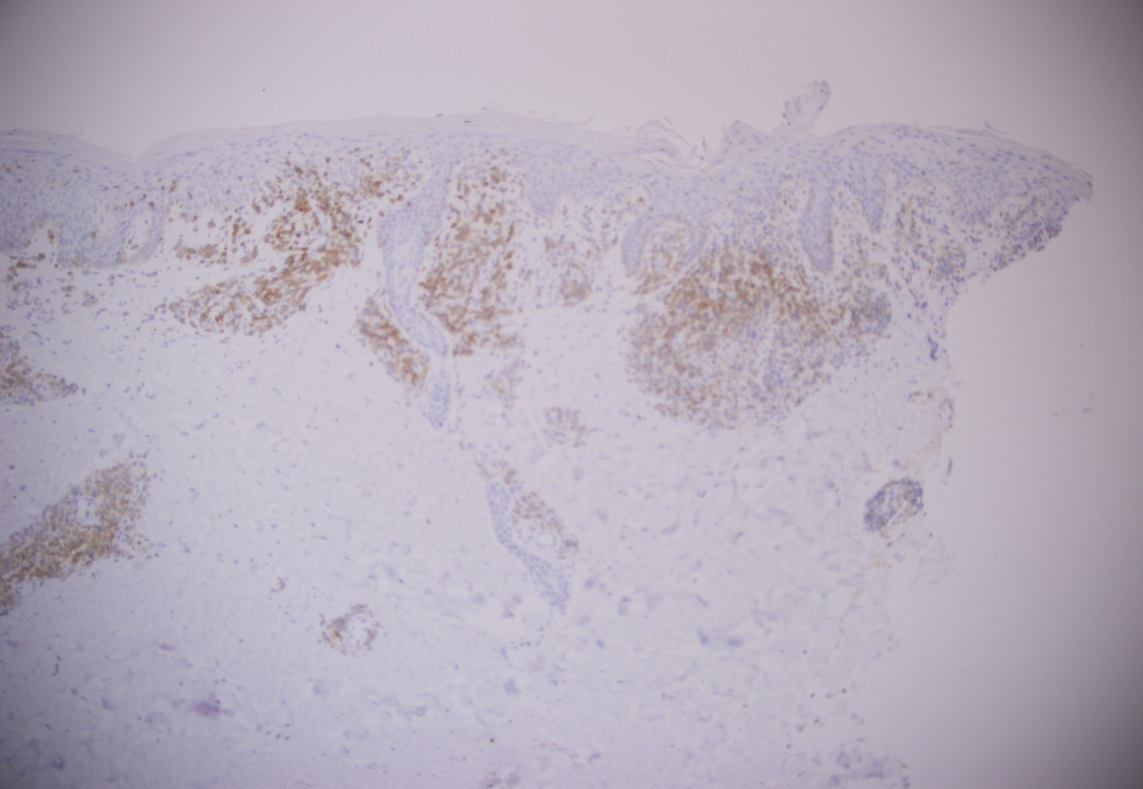 |
|
Figure 7 |
|
|
|
Figure 8 |
|
|
|
Figure 9 |
|
|

References
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
- HSI, E. D. HEMATOPATHOLOGY: foundations in diagnostic pathology. S.l.: ELSEVIER; 2007.
 Meet our Residency Program Director
Meet our Residency Program Director
 LeShelle May
LeShelle May Chancellor Gary May
Chancellor Gary May