
Residency Program - Case of the Month
March 2010 Final Diagnosis - Presented by Melissa Rodgers-Ohlau, M.D.
Answer:
Metastatic Adrenal Cortical Carcinoma
Histologic description:
Microscopic examination of the ovarian mass revealed large nests of cells interspersed with clefts or sinusoids (Figures 2 & 3.) Large areas of necrosis were seen as well (Figure 2). Cells contained pleomorphic hyperchromatic nuclei, often large and bizarre in appearance or even multinucleated. The cytoplasm was eosinophilic and bubbly; cell borders were predominantly indistinct. Rare atypical mitotic figures were also noted (Figure 4). The omental masses showed similar features, though the nuclei were even more bizarre, containing vesicular chromatin and prominent nucleoli (Figures 5 & 6).
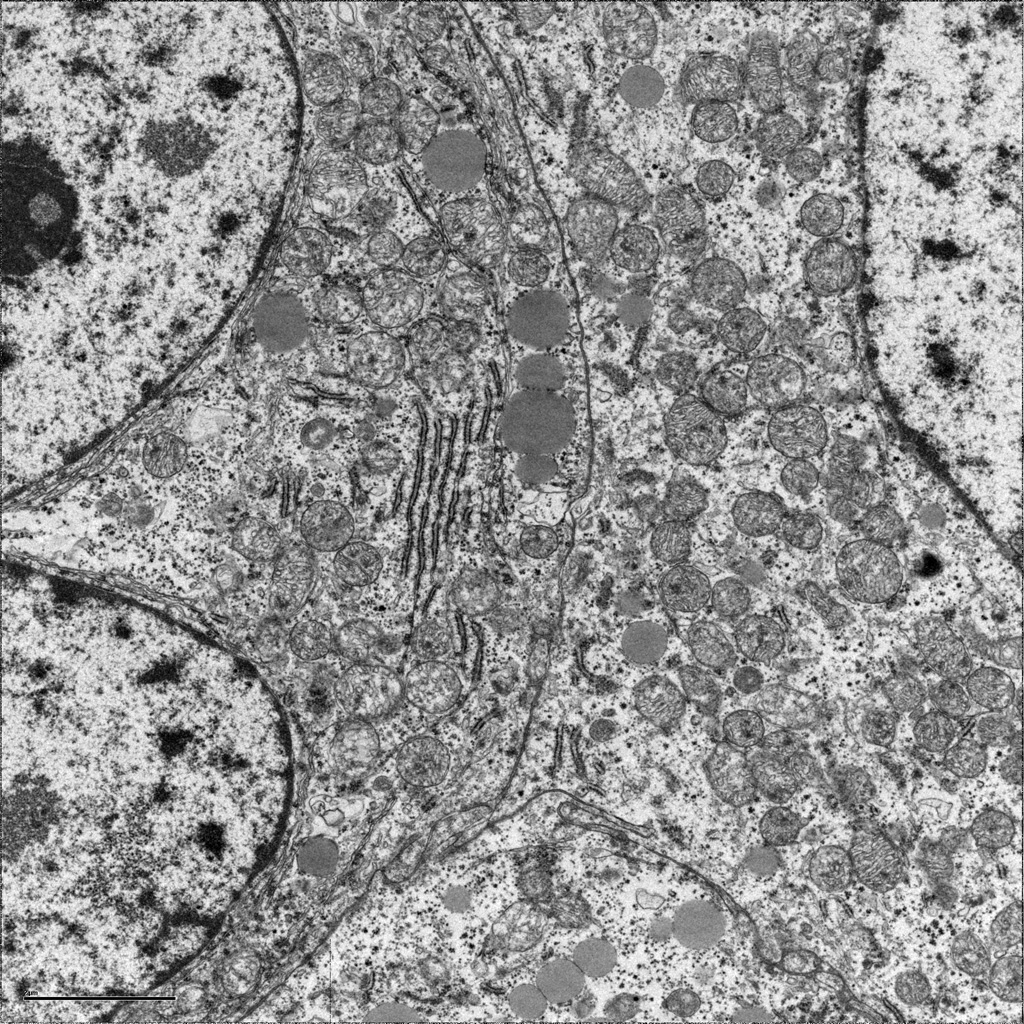
Ultrastructural examination by electron microscopy revealed cells with short stacks of rough endoplasmic reticulum, abundant lipid droplets and possible intramitochondrial inclusions.
Investigation of the patient's past medical history revealed that the patient had a left adrenalectomy 10 years prior. The patient could provide very few details about this but was told the adrenal gland was enlarged but not malignant; she received no chemotherapy or radiation. Medical records concerning this past medical history could not be obtained..
Discussion:
For this case the differential diagnosis included ACC, malignant pheochromocytoma, poorly differentiated tumor of ovarian origin and poorly differentiated carcinoma.
| Differential Diagnosis | Morphology | Immunohistochemistry |
| Adrenal cortical carcinoma | May see sheets of cells, trabeculae or serpiginous cords or nests Cells with eosinophilic, bubbly Cytoplasm; varaiable nuclear Atypia, possible bizarre hyperchromatic nuclei; mitotic figures – variable (some atypical) |
Positive: Inhibin, Melan-A, bcl-2, Synaptophysin (variable), S-100 (variable), vimentin, Cam5.2 (variable) Negative: chromogranin, AE1/AE3 (negative to weakly positive), EMA, CEA |
| Malignant pheochromocytoma | Zellballen architecture or trabeculae Cellular monotony to profound Pleomorphism; relatively small Cells with high N:C ratio; nuclear hyperchromasia with prominent nucleoli; increased mitoses |
Positive: Chromogranin; synapto physin (variable); vimentin (variable), S-100 (variable) Negative: Inhibin, Melan-A, keratin (usually), calretinin |
| Poorly differentiated ovarian tumor: | ||
| Germ cell tumor | ||
| Choriocarcinoma | Mixture of syncytial and cytotrophoblastic elements with hemorrhage and necrosis | Positive: hCG, CD10, HPL, EMA (syncytiotrophoblast),Cytokeratin, CEA (variable) |
| Dysgerminoma | Nests of tumor cells separated by fibrous stroma with lymphocytes | Positive: OCT-4, CAM 5.2 (variable), AE1/AE3 (variable) Negative: EMA, HMW keratin, vimentin |
| Embryonal | Sheets and nests of large primitive cells, syncytiotrophoblast cells may be seen as well | Positive: hCG 9if syncytiotrophoblast is present), cytokeratin, PLAP, CD30 Negative: EMA, mucin |
| Carcinoid | Insular or trabecular pattern resembles carcinoid elsewhere in body, rarely has prominent pleomorphism |
Positive: Chromogranin, synaptophysin, NSE Negative: Inhibin |
| Sex cord tumor | ||
| Granulosa cell tumor | Bland cuboidal to polygonal cells in various patterns; cells may be plump with abundant cytoplasm; cells with coffee bean nuclei with folds | Positive: Inhibin, vimentin, calretinin, SMA, keratin (variable) Negative: EMA |
| Thecoma | Spindle cells with moderate pale cytoplasm may conatin lipid droplets; centrally located nuclei; intervening stroma with collagen | Positive: Oil red O stains lipid droplets, reticulin is noted to surrounded clusters of cells |
| Poorly differentiated carcinoma | Variable | Variable |
The patient's remote history of adrenalectomy, combined with the gross and microscopic findings led us to heavily favor an adrenal cortical origin for this tumor, with ACC being the most likely diagnosis. IHC staining and EM findings allowed us to rule out a tumor originating in the adrenal medulla and confirmed our suspicion that this tumor was a metastatic adrenal cortical carcinoma.
The incidence of adrenal cortical carcinoma (ACC) is 0.1 cases in 100.000, making up approximately 3% of all endrocrine neoplasms. There is some disagreement over sex predilection, with most experts agreeing that men and women are equally affected. Adults are most commonly affected, usually in the fifth to sixth decade of life, but pediatric cases have been reported, often in association with specific genetic syndromes (discussed later). [1, 6, 8]
The most common presenting symptoms of adrenal cortical carcinoma are related to glucocorticoid, mineralcorticoid or androgen excess; however, some tumors are nonfunctioning. ACC may also be detected due to mass effect. These tumors tend to be large – usually greater than 5-6 cm and weighing >50 grams (sometimes more than 1 kilogram) – producing localized symptoms such as flank pain. Highly necrotic tumors may produce fever. [1, 6, 8]
Grossly, the cut surface of an adrenal cortical carcinoma has a variegated appearance with solid homogenous yellow components interspersed with areas of soft, friable nodules and areas of hemorrhage and necrosis (Figure 1). Tumor invasion of major veins is a frequent finding and often leads to total occlusion, thrombosis and embolism. [1, 8]
Microscopically, the tumor may display a variety of architectural patterns, including: 1)well-differentiated, resembling normal adrenal cortex; 2) patternless sheets of cells interrupted by a fine sinusoidal network; 3) broad or fine trabeculae or serpiginous cords of cells; and 4) large nests of cells (Figures 2-6). Other histological findings include fibrous bands, myxoid change and invasion of the capsule and large veins (very common). [1, 8]
Cytological findings include: 1) cells with eosinophilic, bubbly cytoplasm ; 2) nuclear atypia which varies from non-existent to highly pleomorphic; 3) bizarre, hyperchromatic nuclei – often multiple; 4) vesicular nuclei; and 5) mitotic figures which vary from rare to several per HPF with atypical forms often noted (Figures 4 & 6). [1, 8]
Immunohistochemical staining with Inhibin (Figure 7) and Melan-A (Figure 8) is sensitive but not specific for ACC. Chromogranin is negative (Figure 9), which is a very reliable way of distinguishing an adrenal cortical tumor from one originating in the medulla (chromogranin would then be positive). Unlike most epithelial tumors, ACCs are negative to weakly positive for cytokeratin, and negative for EMA and CEA. [1, 8]
Electron microscopy will show cells with abundant rough and smooth endoplasmic reticulum, numerous irregularly-shaped mitochondria which often show cavitation and loss of cristae. The number of lipid droplets (typical of steroid-producing cells) may be abundant to markedly decreased. [1, 12]
CT or MRI typically demonstrates a large mass (> 5 cm) with uneven enhancement, often containing areas of decreased enhancement due to hemorrhage or necrosis. Adjacent organs are frequently displaced or involved by tumor. Invasion of the renal vein and IVC can lead to metastases to the right atrium as well as the lung. ACC most commonly metastasizes to the liver, lung, kidney, peritoneal and pleural surfaces, retroperitoneal lymph nodes and bone. However, any site may be affected, including the skin. While rare, there are reports in the literature of ACC metastasis to the ovary. [1, 3, 8]
With regard to genetic alterations, no specific chromosomal abnormalities have been associated with ACC. Amplifications and losses of specific gene loci have been identified through comparative genomic hybridization, but the patterns are varied and, again, nonspecific. The most frequent molecular genetic alterations associated with ACC involve IGF2, TP53, EGFR and MENIN. While these alterations are most commonly sporadic, there are specific genetic syndromes in which development of adrenal cortical carcinoma is more likely. These include: Beckwith-Wiedemann syndrome in which paternal uniparental isodisomy leads to overexpression of IGF2; Li-Fraumeni syndrome in which patients have germline mutations in TP53; and MEN I, in which patients have germline mutations in the MENIN gene. [1, 8, 9, 10]
There is no currently accepted staging system for adrenal cortical carcinoma. The staging system most commonly used, developed by McFarlane and modified by Sullivan, is based on size of the primary tumor, degree of local invasion, regional lymph node involvement and distant metastases (See Table 1). [1, 4, 5, 7, 11] Currently, Stage IV includes both potentially resectable locally advanced disease and distant metastases. However, patients without distant metastases fare better than those with distant metastases; therefore, it has been proposed that Stage IV be changed to include only those patients with distant metastases. [2] Based on a study by Fassnacht, et al. [2], this would change the staging prognosis for ACC, with regard to 5-year survival rates, as follows:
| Old | New | |
| Stage I | 82% | 82% |
| Stage II | 58% | 61% |
| Stage III | 55% | 50% |
| Stage IV | 18% | 13% |
Currently, the majority of cases present with Stage IV disease, with approximately 40% of cases presenting with distant metastases. In addition to tumor stage at diagnosis, patient age (pediatric patients usually have a better prognosis), mitotic index, tumor size and Ki-67 index (usually between 5-20%) are important prognostic indicators. [1]
Treatment for ACC is primarily surgical excision. These tumors have poor radiosensitivity but do respond to chemotherapy with mitotane, either in an adjuvant role, as primary treatment in unresectable tumors or in situations in which ACC recurs. [1]
References:
- DeLellis, Ronald A., Lloyd, Ricardo V., Heitz, Philipp U. and Eng, Charis. (Eds.): WHO Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. IARC Press: Lyon 2004.
- Fassnacht M; Johanssen S, et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009 Jan 15;115(2):243-250.
- Kurek R, Von Knobloch R, Feek U, et al. Local recurrence of an oncocytic adrenocortical carcinoma with ovary metastasis. J Urol. 2001 Sep;166(3):985.
- MacFarlane, DA. Cancer of the adrenal cortex; the natural history, prognosis and treatment in a study of fifty-five cases. Ann R Coll Surg Engl 1958; 23:155.
- National Cancer Institute. Adrenocortical Carcinoma Treatment (PDQ®). Stage Information. http://www.cancer.gov/cancertopics/pdq/treatment/adrenocortical/HealthProfessional/page4.
- Ng, L and Libertino, JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol 2003 Jan;169(1):5-11.
- Norton, JA, Le, HN. Adrenal tumors. In: Principles and Practice of Oncology (DeVita Jr, VT, Hellman, S, Rosenberg, SA eds), 7th ed, Lippincott, Williams, and Wilkins, Philadelphia, 2005. p 1534.
- Rosai, Juan MD. Rosai and Ackerman's Surgical Pathology. Volume One. Ninth Edition. New York: Mosby, 2004. 1119-1139 and 1649-1736.
- Rosati R, Cerrato F, Doghman M, et al. High frequency of loss of heterozygosity at 11p15 and IGF2 overexpression are not related to clinical outcome in childhood adrenocortical tumors positive for the R337H TP53 mutation. Cancer Genetics and Cytogenetics. Volume 186, Issue 1, October 1, 2008. Pages 19-24.
- Schneider K. and Li F., "Li Fraumeni Syndrome." Gene Reviews. http://www.geneclinics.org/profiles/li-fraumeni/details.html.
- Sullivan M; Boileau M; Hodges CV. Adrenal cortical carcinoma. J Urol 1978 Dec;120(6):660-5.
- Trump, Benjamin F., Jones, Raymond T. Diagnostic Electron Microscopy. Volume 3. John Wiley & Sons, New York, 1980.
|
Figure 1
|
Figure 2
|
|
Figure 3
|
Figure 4
|
|
Figure 5
|
Figure 6
|
|
Figure 7
|
Figure 8
|
|
Figure 9
|









Table
| Tumor (T) | |||
| T1 | Tumor 5 cm or less in greatest dimension, local invasion absent | ||
| T2 | Tumor more than 5 cm in greatest dimension, local invasion absent | ||
| T3 | Tumor outside adrenal, in fat | ||
| T4 | Tumor invading adjacent organs | ||
| Regional lymph nodes (N) | |||
| N0 | No regional lymph node metastasis | ||
| N1 | Regional lymph node metastasis | ||
| Distant metastasis (M) | |||
| M0 | No distant metastasis | ||
| M1 | Distant metastasis present | ||
| Stage grouping | |||
| Stage I | T1 | N0 | M0 |
| Stage II | T2 | N0 | M0 |
| Stage III | T1-2 | N1 | M0 |
| T3 | N0 | M0 | |
| any T | any N | M1 | |
| Stage IV | T3 | N1 | M0 |
| T4 | N0 | M0 | |
 Meet our Residency Program Director
Meet our Residency Program Director
 LeShelle May
LeShelle May Chancellor Gary May
Chancellor Gary May